近日,我院多尺度模拟计算科研团队在化学工程领域顶级期刊Chemical Engineering Science上发表了题为“Fe3cluster-anchored monolayer MoS2for direct deoxygenation of phenol: catalyst design and activation mechanism”的研究论文。论文第一作者为2022级材料化工专业硕士研究生马佳丽同学,通讯作者为王鑫博士和刘兴满博士。
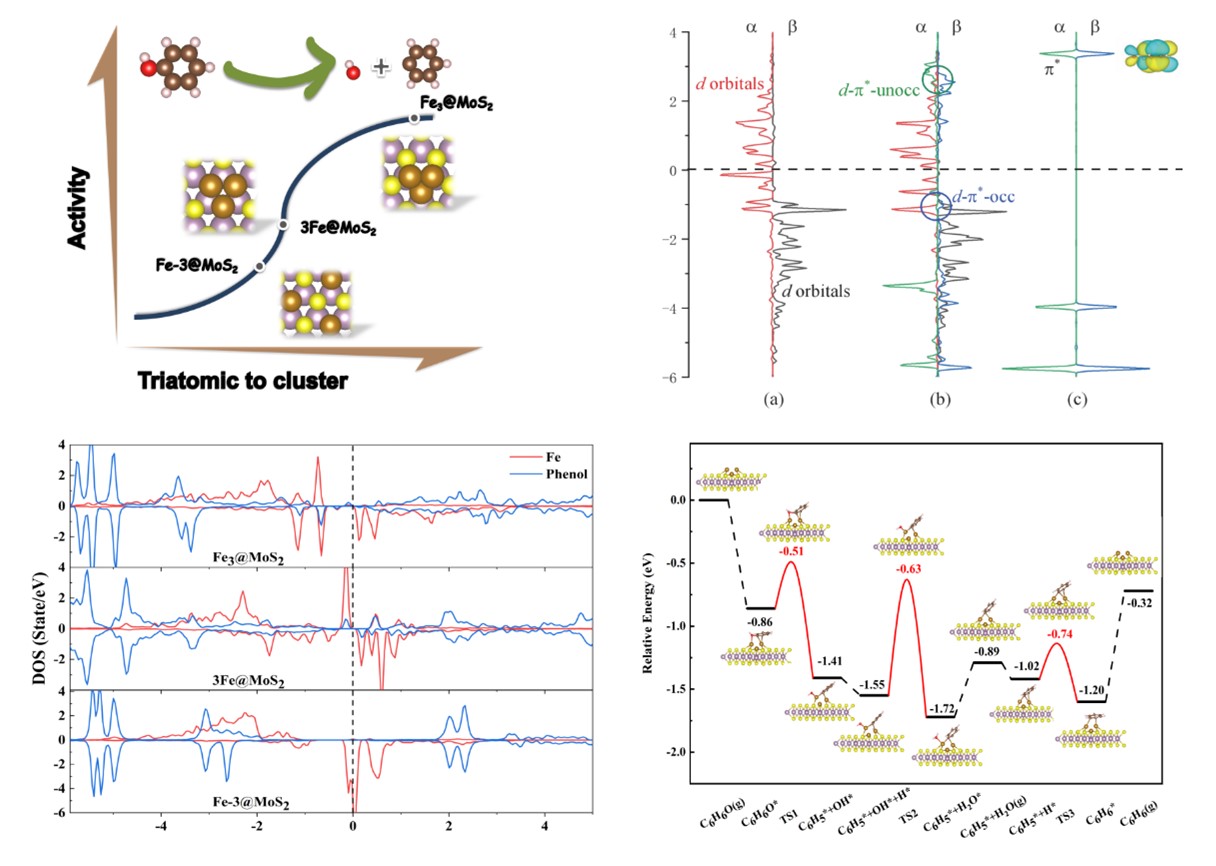
本研究中,研究人员创新性地设计了Fe3@MoS2催化剂,并通过密度泛函理论(DFT)计算深入剖析了Fe3团簇上C-O键直接断裂的电子转移和活化能垒。他们发现,一种独特的活化机制在此过程中发挥了关键作用,即d-π轨道的形成与占据,这一机制极大地促进了电子从Fe3的d轨道向酚类C-O键的π轨道转移,从而显著降低了反应能垒。
进一步的研究表明,Fe3@MoS2催化剂的卓越催化活性源自Fe3团簇的自旋极化和较低的氧化态,这一发现得到了态密度(DOS)和Bader电荷分析的有力支持。这些特性使得Fe3@MoS2在催化酚类直接脱氧(DDO)反应中展现出更高的活性和选择性。
该研究成果不仅为木质素的高效转化提供了新的催化剂选项,也为理解多相催化过程中的电子转移和活化机制提供了新的视角。
文章链接:https://authors.elsevier.com/a/1je7K26dc2XS1





 学校主页
学校主页